Junk in the (nr) Trunk
You often find the darndest things if you just look!
As a required task for parts of my thesis project, I looked at every Papillomavirus (PV) sequence in the NCBI Virus database to see if there were any integration sites, or other sequence contamination. This can be done in a few ways, which boils down to “are there any regions in our PV sequences that align unexpectedly well to things that are not PVs?”.
One of the ways is taking all the PV sequences, and running an alignment against some genome of your choice. For example, PV seqs vs. E. coli genome. If there are any high confidence alignments between these two, you can be reasonably sure that for some reason or the other, there are bacterial sequences in your PVs. I wrote a slightly buggy script that does this here.
Obviously the above method is far from perfect - you can run a set of genomes that are commonly found as contaminants, like human, mouse or yeast, but, you can never truly capture all the possible contaminants and genomes from which they come from. So to band-aid this a little, what you can do it align the PV sequences to a small subset of random libraries from Logan (3400, provided by Rayan). If there are any regions that align to a lot of things, then that gives you a starting point of where to look. Unfortunately the actual inspection will need to be manual!
Visual example of the “hotspot” method:
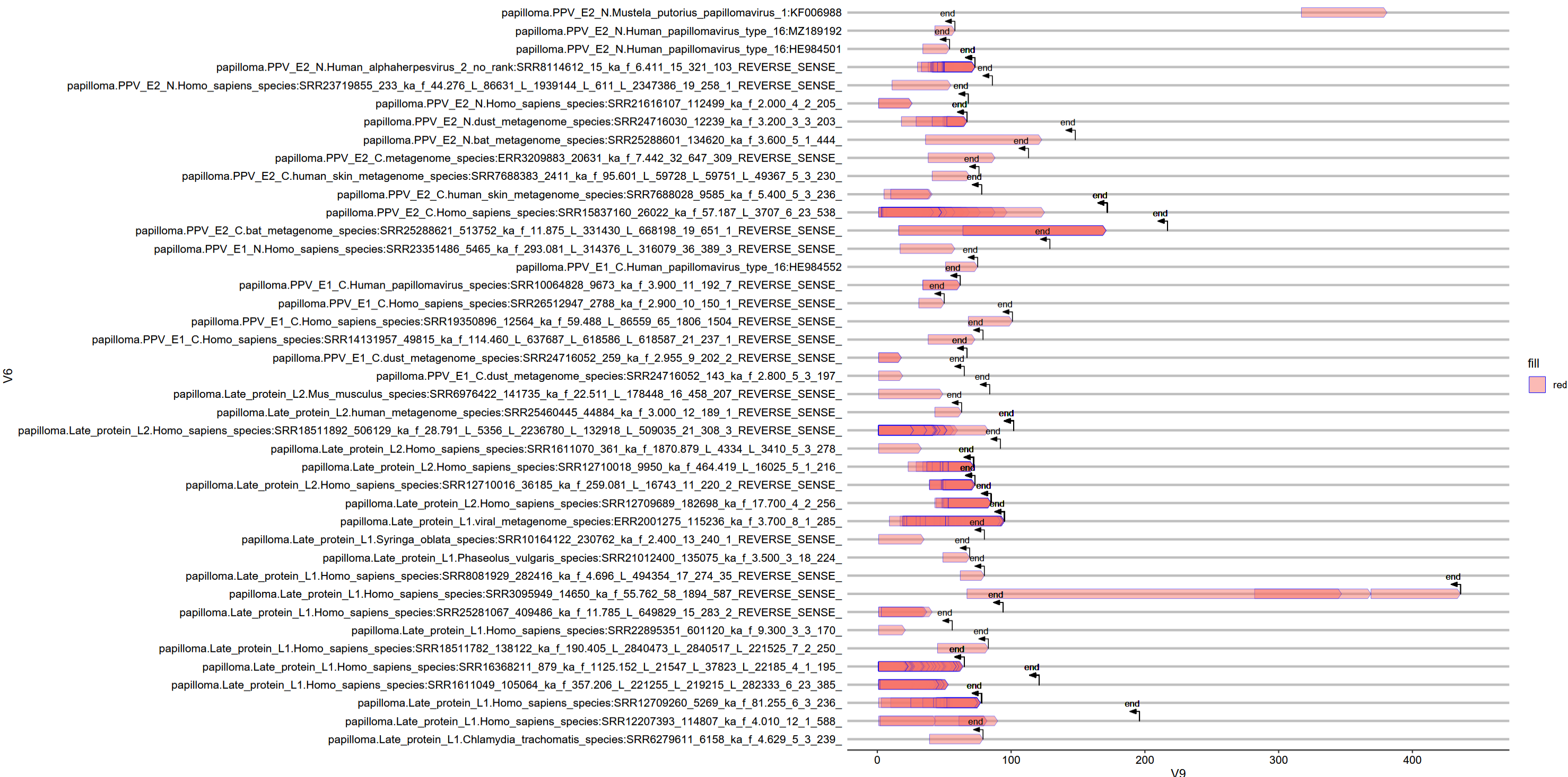
Most of the time, what I found was a ton of human genome integration sites - this is expected, lots of people do studies on when, where, and how PVs integrate into the human genome. After all, it is one of the main risk factors for and during development of PV-associated cancers.
But some of the time, there were some interesting things!
Part 1: Bacteria in the bovine
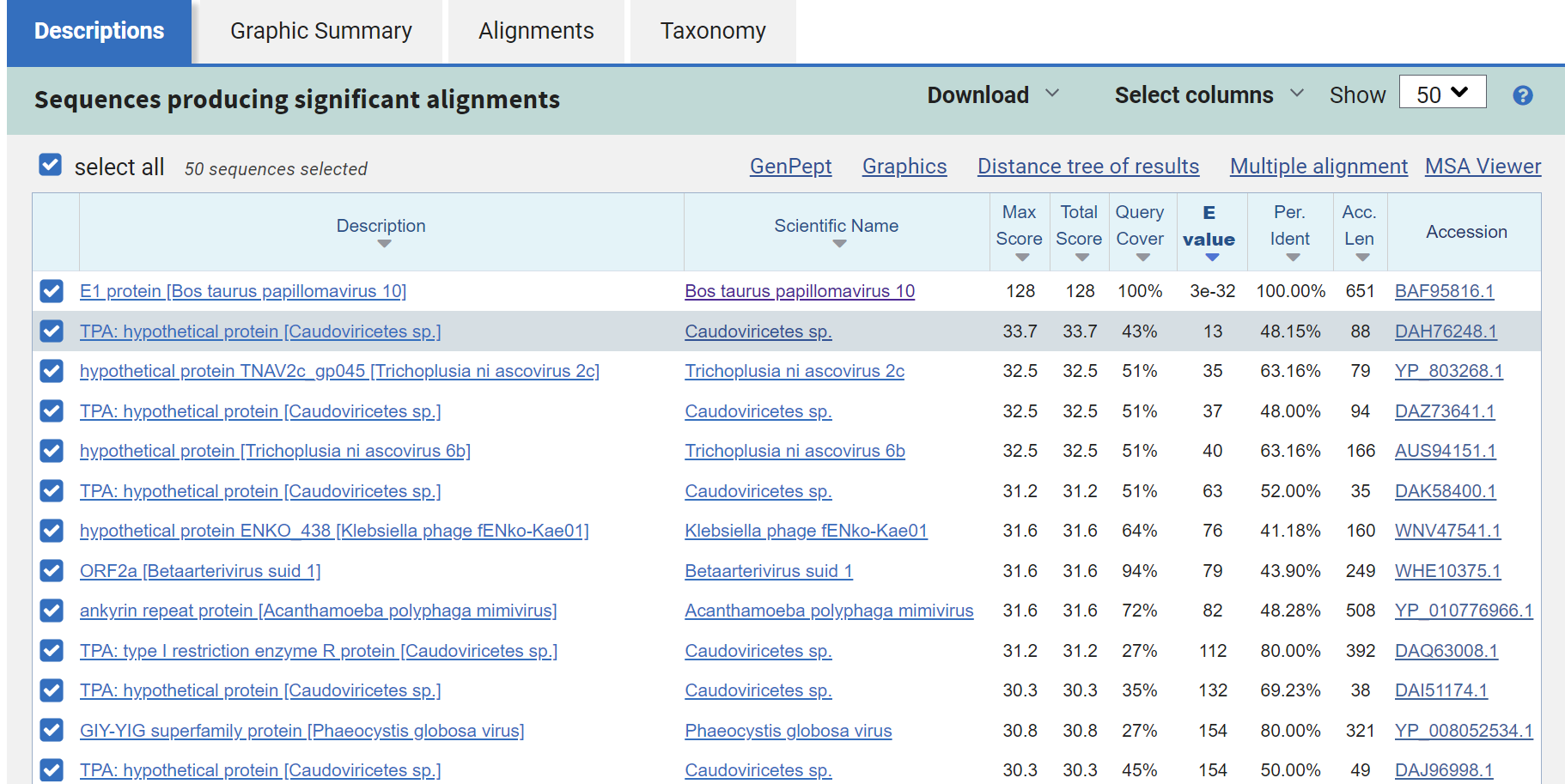
One of those was an entry in the BLASTnr database (Acc: AB331651), of the E1 replication protein of a bovine papillomavirus. Upon investigation of “hotspots” (using a slightly different visualization method because I was in a rush), you can see that the green circled sequence has a lot more alignments (represented as dots here) compared to the rest of it’s brethren, except for MZ189188. That one is a proviral integration site study.
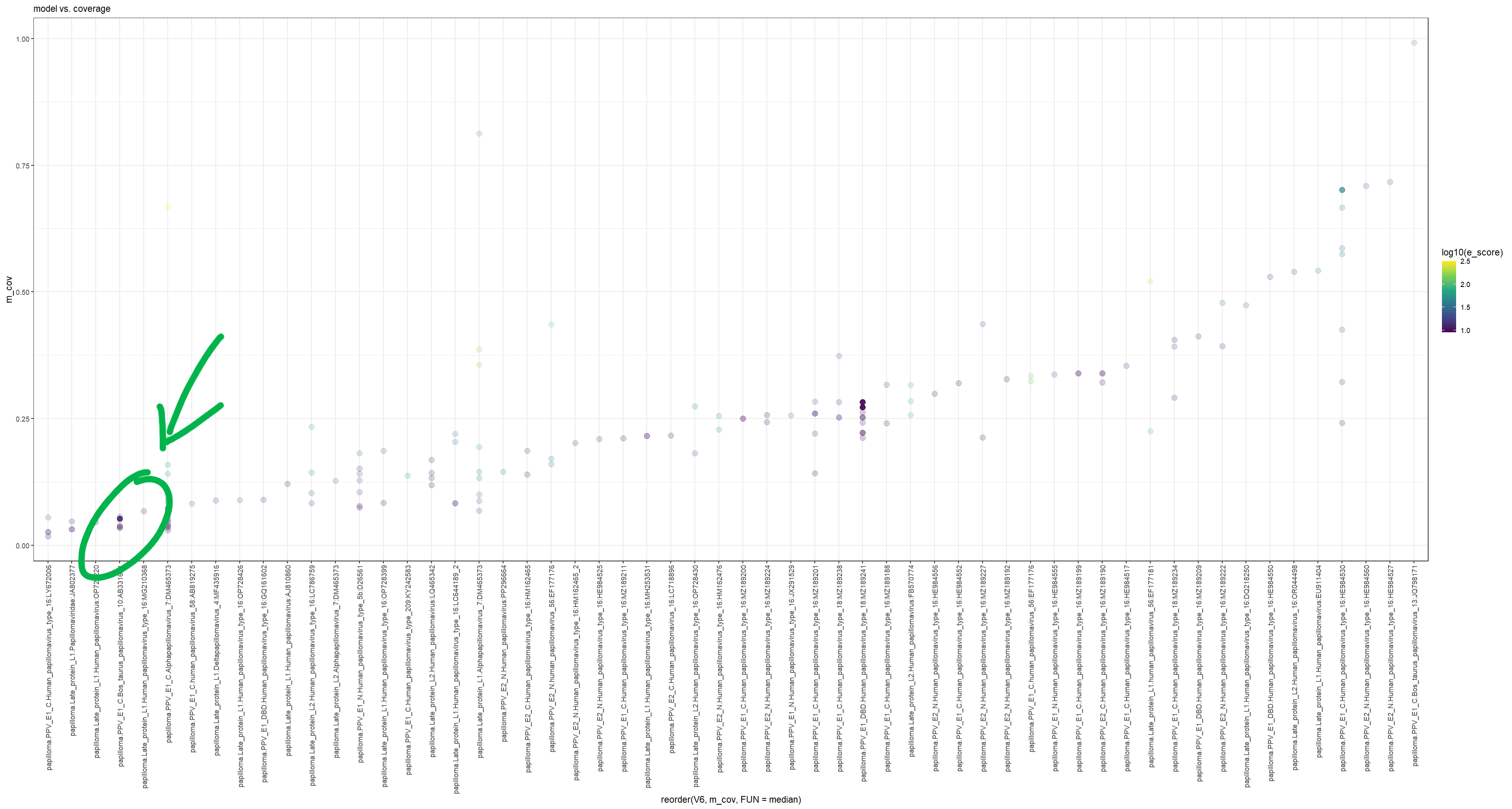
But, AB331651, had this region from amino acids 381-417 that are consistently being hit by all kinds of libraries. Taking this the nucleic acids of Logan contigs that aligned to this sequence, and conducting a BLASTx search, all of these regions were for bacterial proteins, mostly transcription factors, transposases, endonucleases, and hypothetical proteins. Taking the region on the PV sequence hit, same results.
Thats not enough evidence to say that there’s something wrong with this sequence! Sure, it could be that there is an error in this sequence, but it could be mis-annotated bacterial proteins (likely not given the diversity of hits returned), or some kind of biological artifact of homology or integration (this would be an interesting finding). To really untangle this, I did a few more things.
First, is to cast a wider net with HMMs - using the Pfam web portal, you can annotate each region of an input amino acid sequence as whatever domains HMMs recognize it as. For this sequence, the putative bacterial sequence is identified as a zinc finger domain, so it is something possibly functional. Then, is this biological?
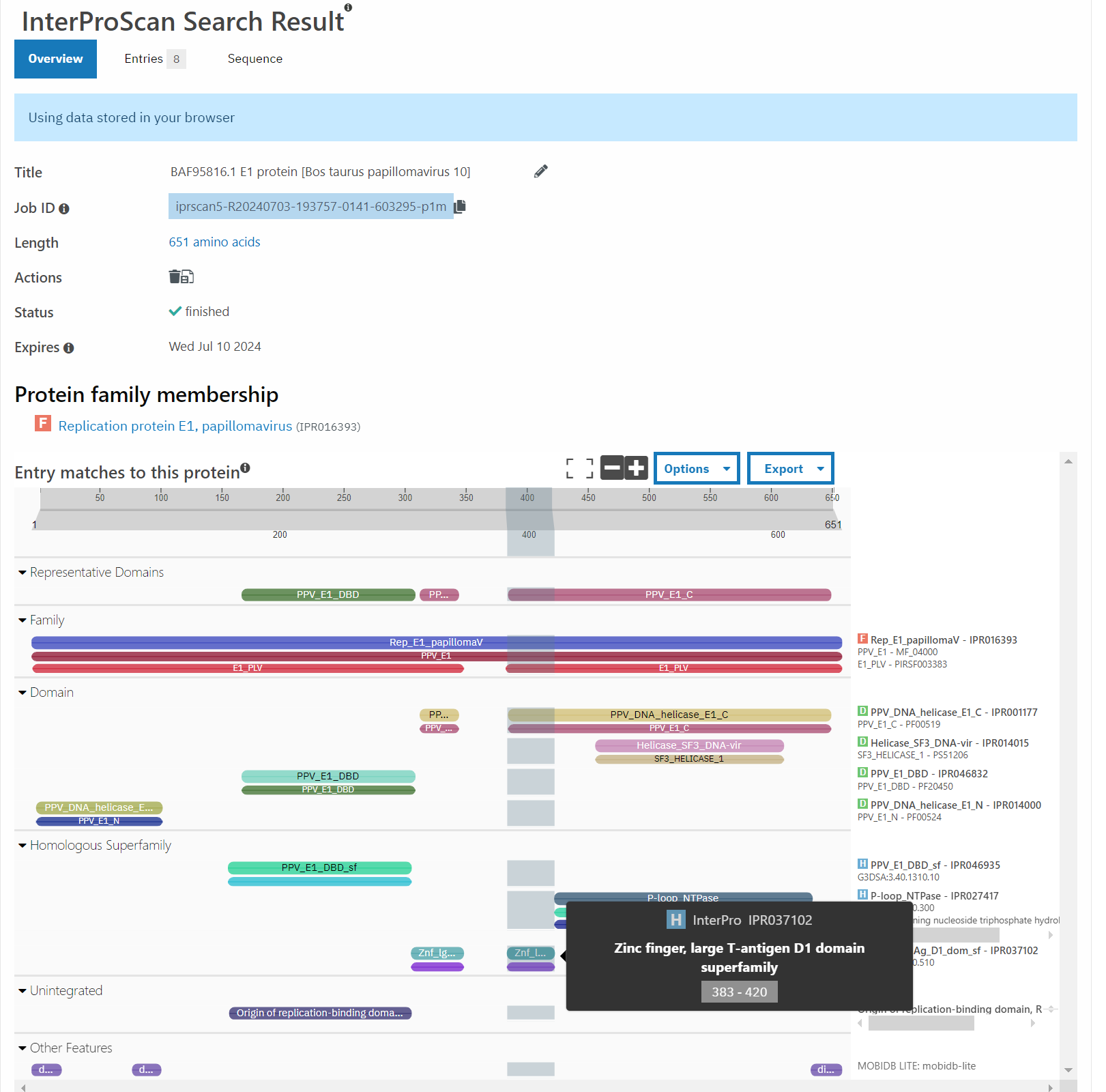
You can only start giving weight to the idea that this is actually relevant to the organism if n > 1, which means that: does this happen literally anywhere else?
A good point to start with this question are things that are most closely related to this bovine PV. According to PaVE, here is the region of the phylogenetic tree that BPV 10 (our AB331651) lives in.
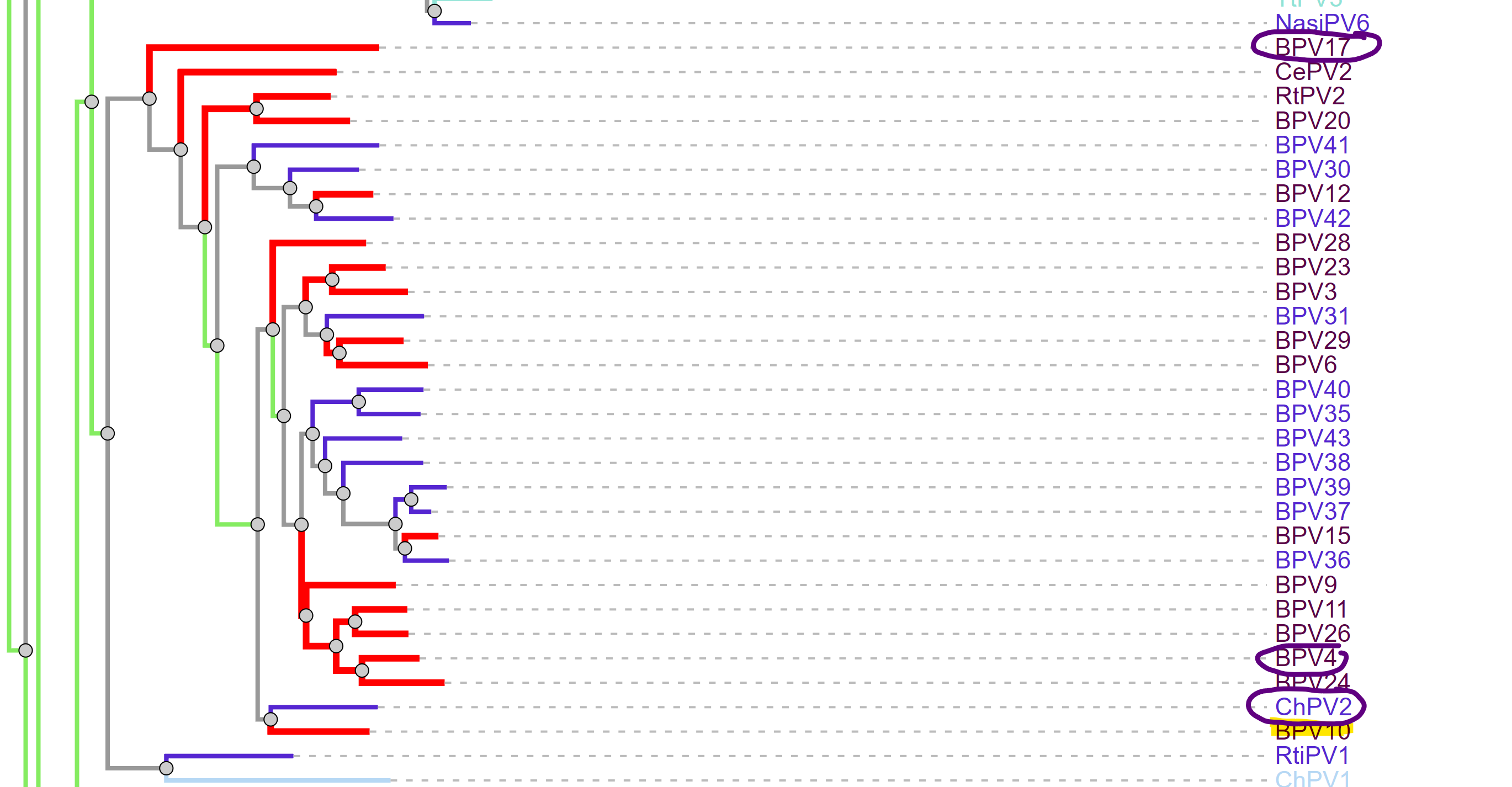
Taking the next closest related PV, ChVP2 (goat), BPV 17 and 4, as well as HPV 18 (for fun), and running those through Pfam, exactly zero of these have the second zinc finger domain.
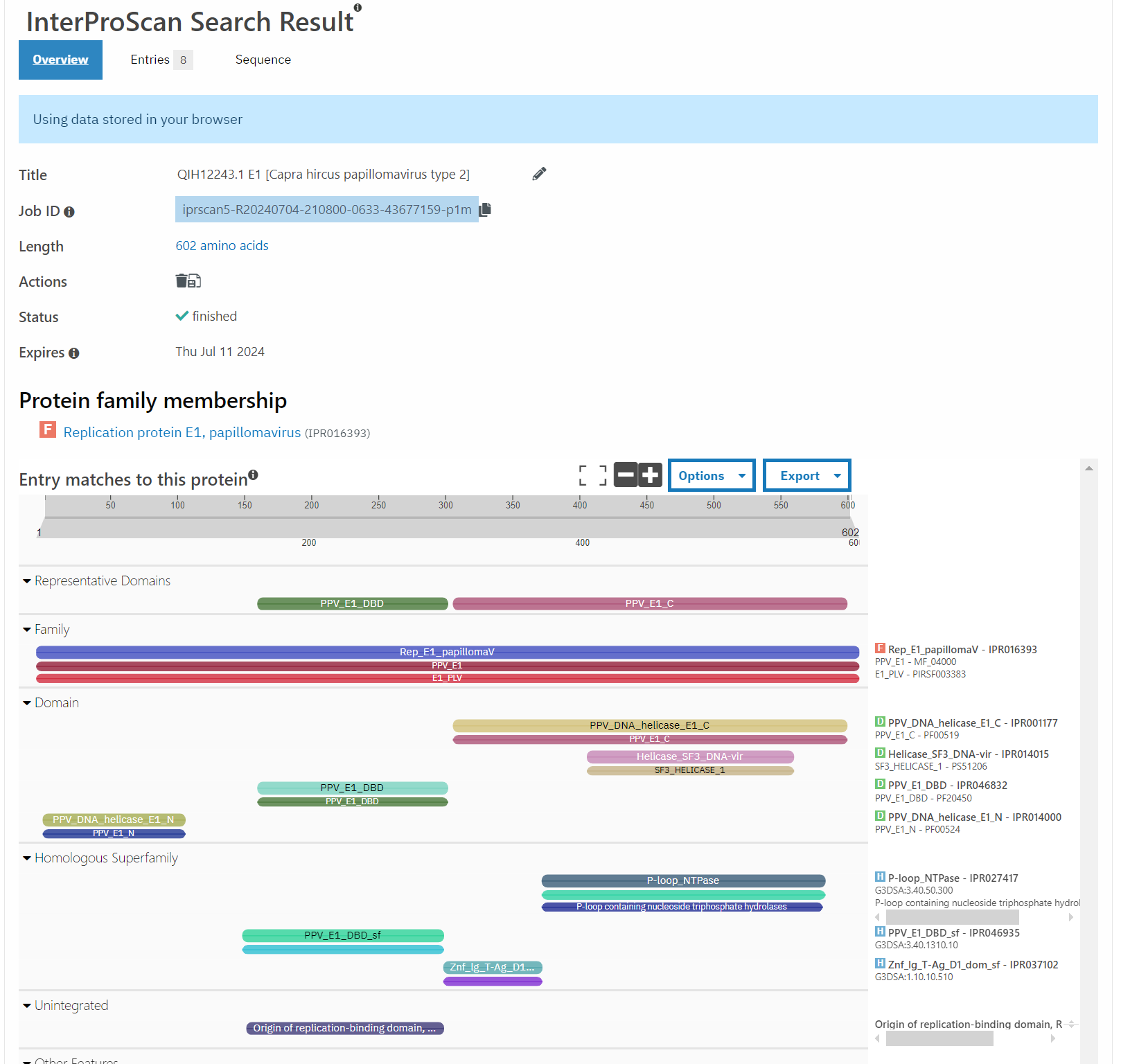
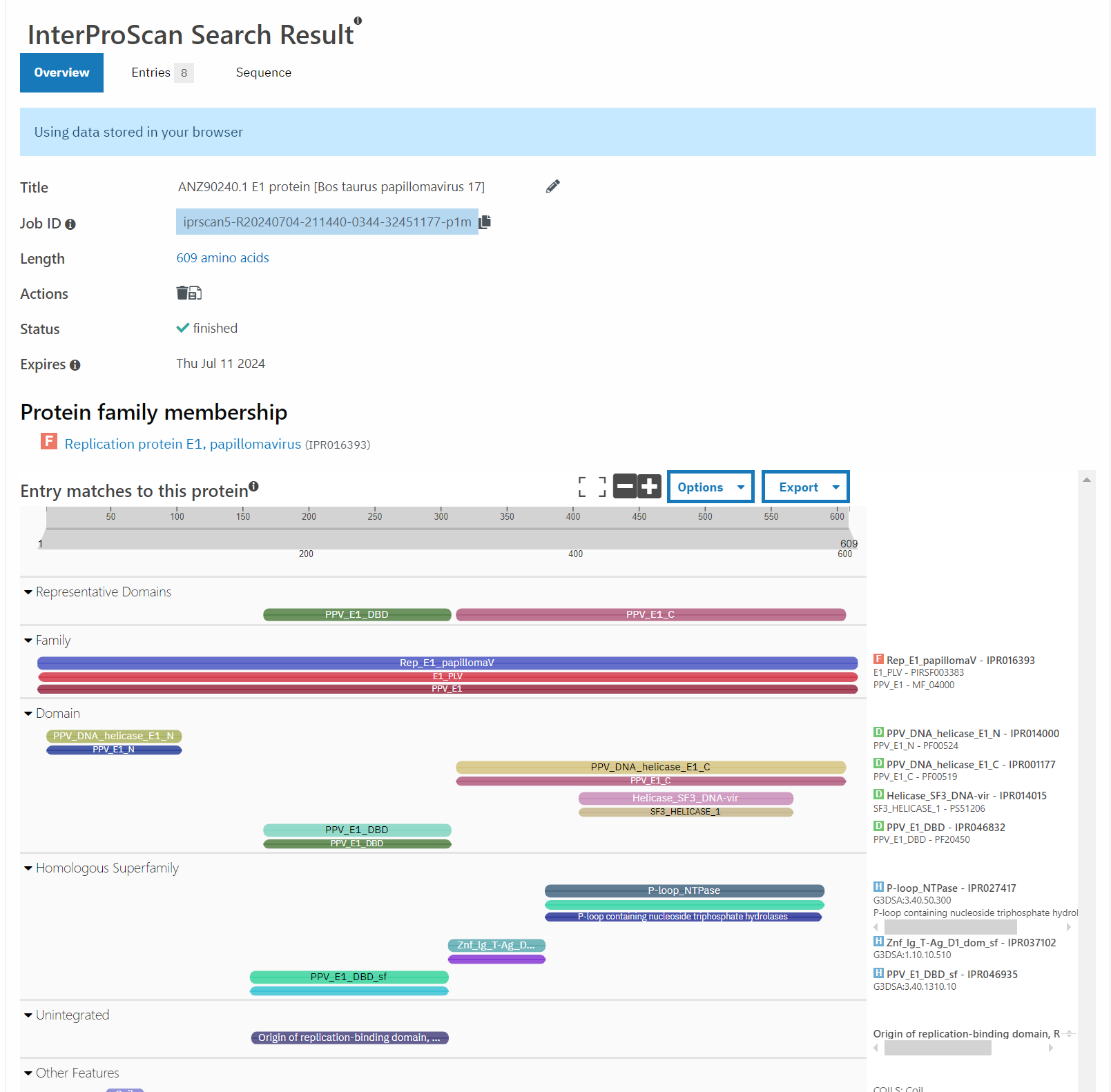
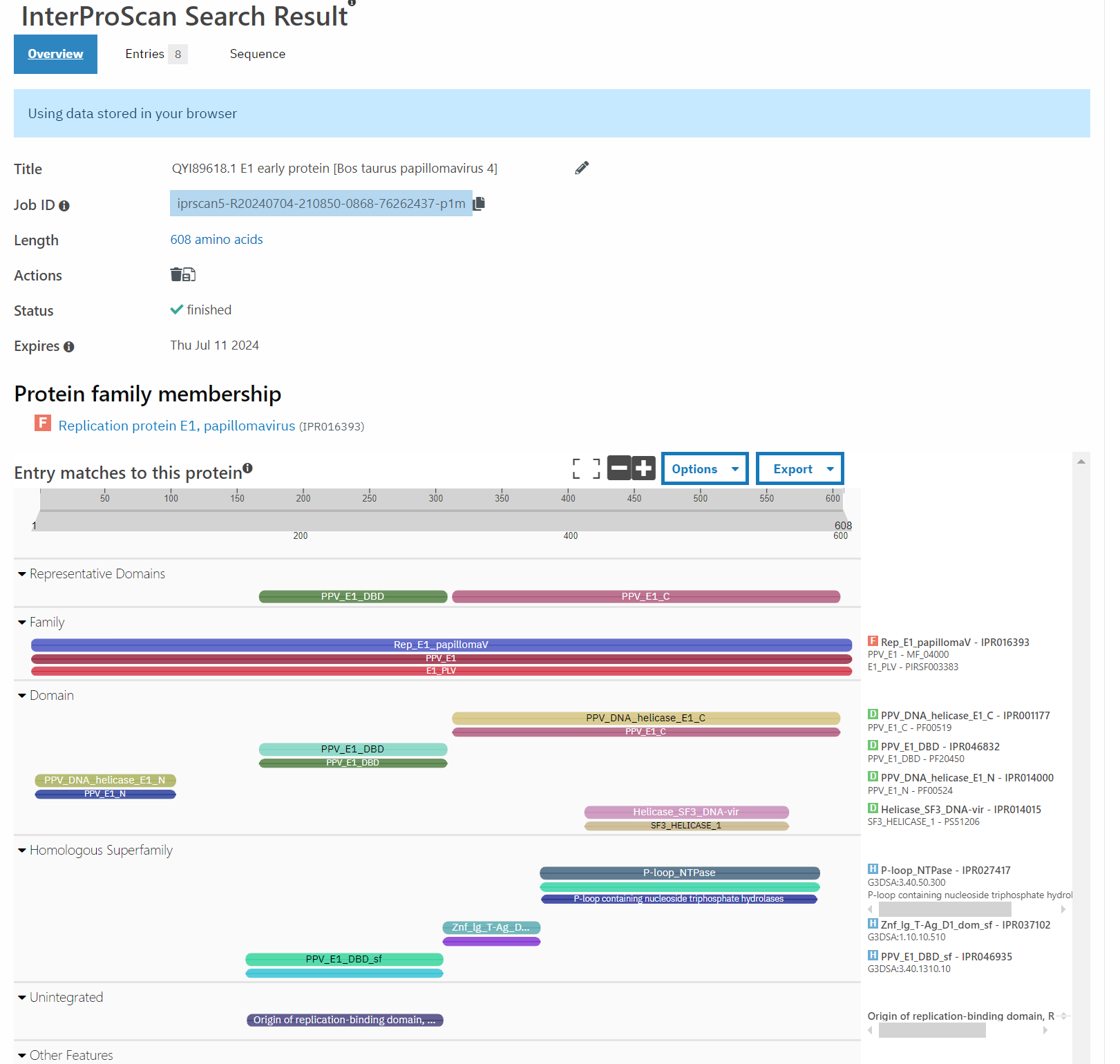
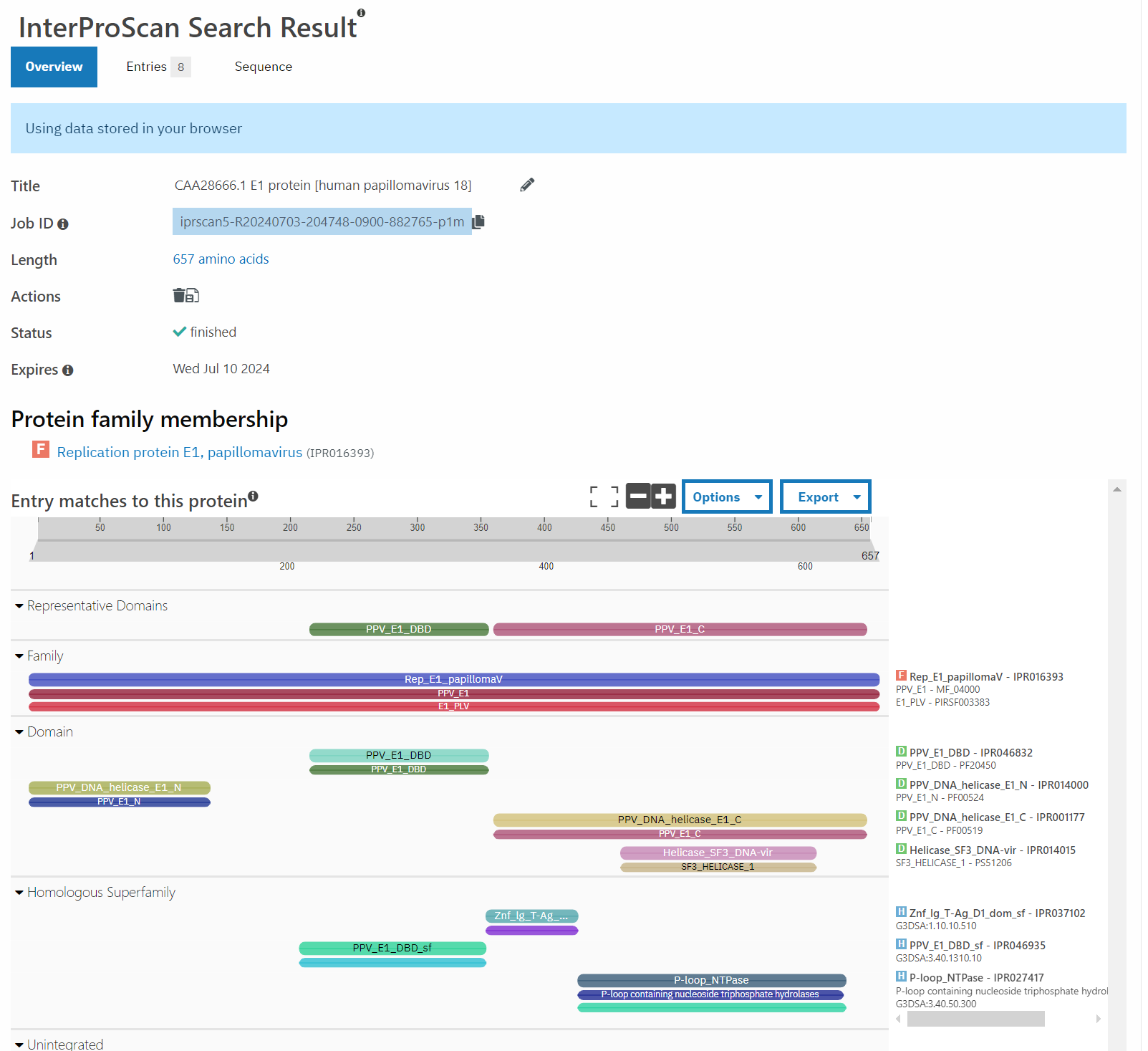
Well, that’s not very promising, is it?
Finally, taking this sequence and running it against all the viruses in the BLASTnr database, it doesn’t exist except for once in this specific BPV 10 sequence, none of the others!
To add injury to insult, no other BPV 10 sequences on NCBI have this second domain/subsequence as well, with annotated with the Pfam database.
n = 1!
In conclusion: This is likely some technical error, or one off event, that was captured by the folks who sequenced AB331651. Since this is the reference BPV10 genome referenced on PaVE, it should probably be remedied, or revisited for posterities sake (I emailed the submitters, but did not receive a response).
Part 2: Mosaics in the murine
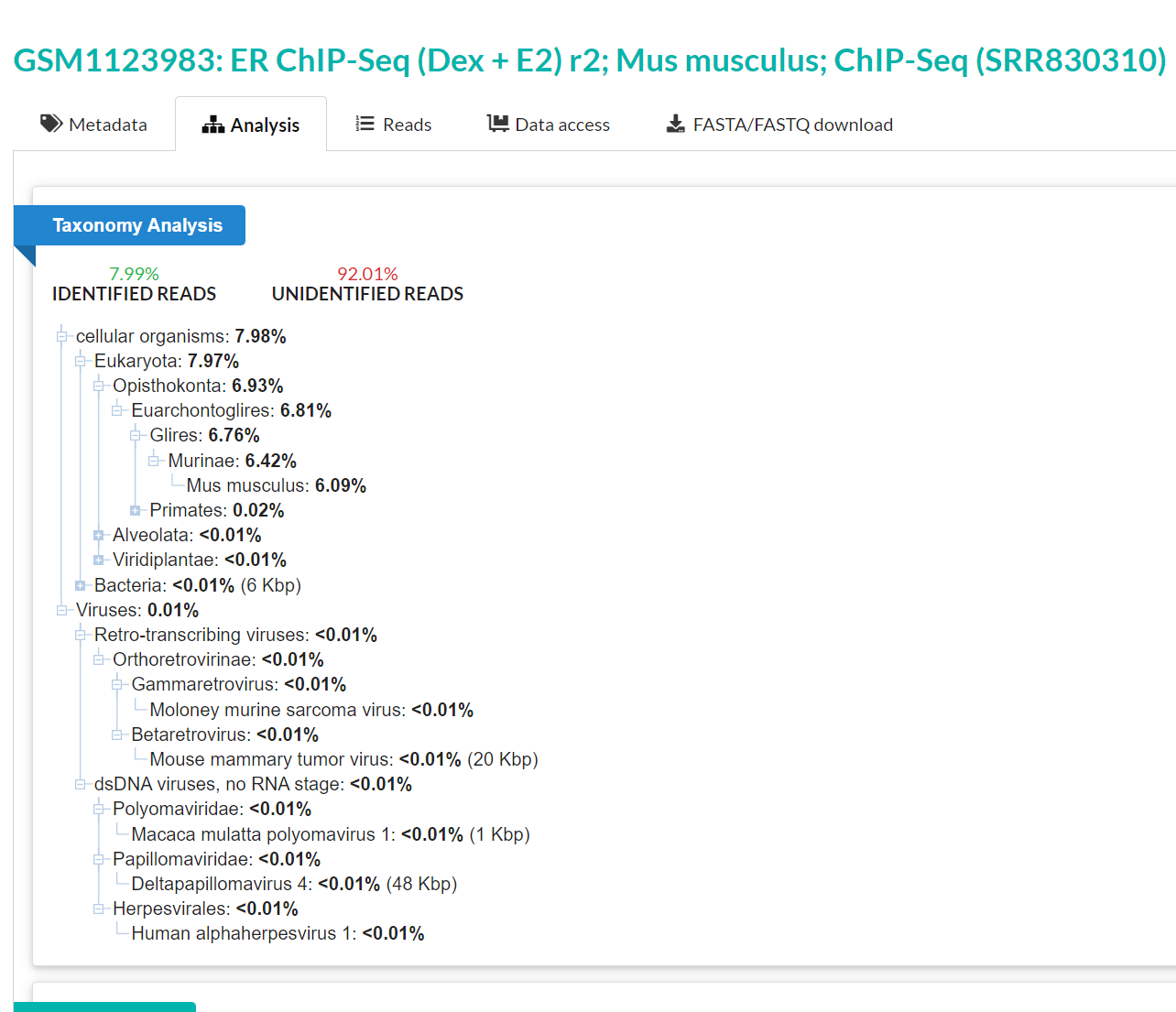
This one was a bit more serendipitous - in the small 3400 accession test set that was used above to look at hotspots, I was interested in seeing if we could get any longer hits. Nothing was a full length L1, and no contigs were >2kb, except for a single one, which drew my attention. This sequence was from library SRR830310, a mouse ChIP-seq library, rather unexpectedly. It’s not impossible that say, there was some slight contamination that resulted in this being assembled, but even the STAT analysis is able to identify there are PV sequences there, meaning that there is a significant amount of reads in there of PV origin.
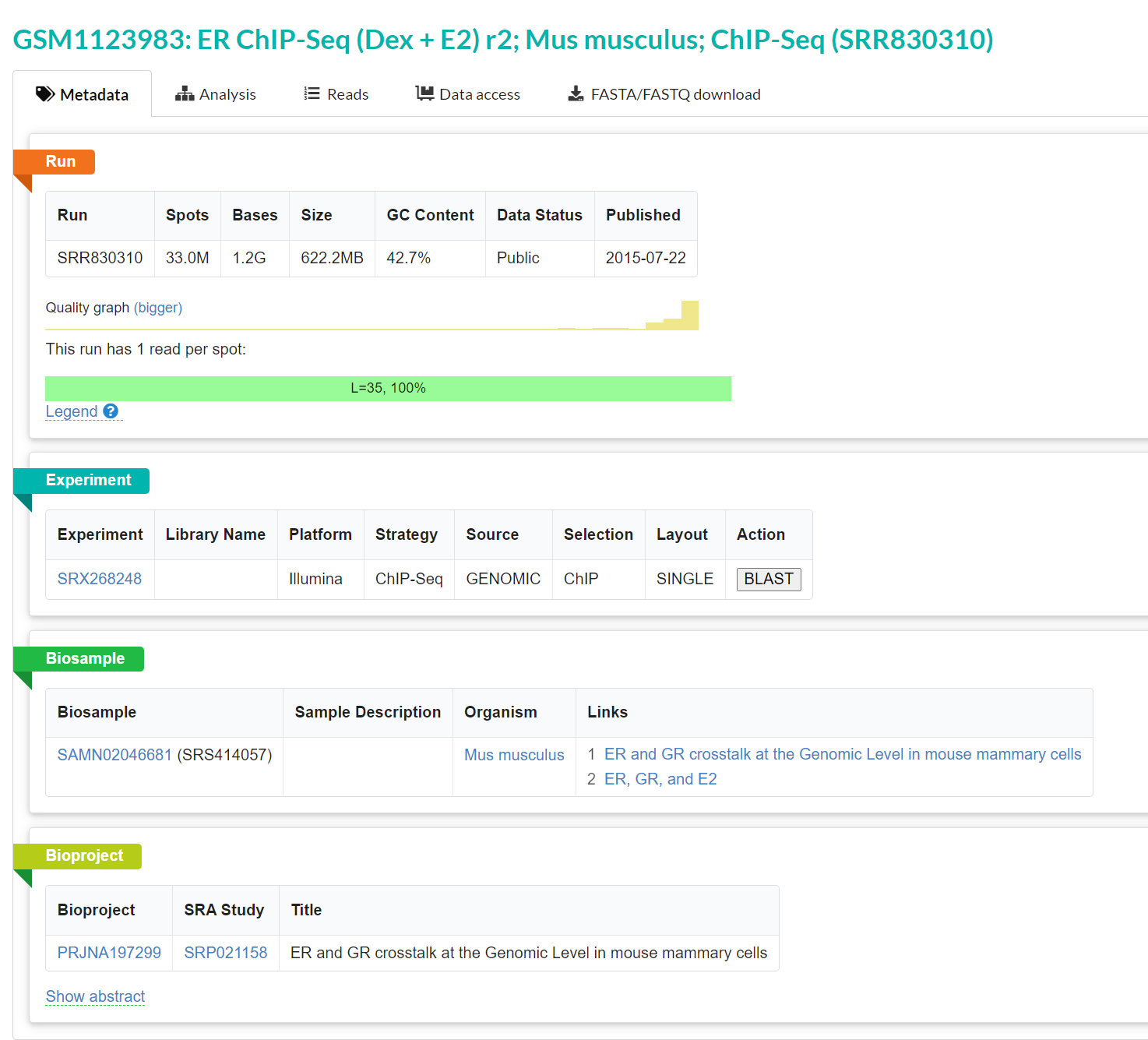
Looking at a single contig, with the same strategy of Pfam in the first scenario, there was surprisingly a (what appeared to be) chimeric contig of PV and also mouse murine tumor virus (MMTV)! What!
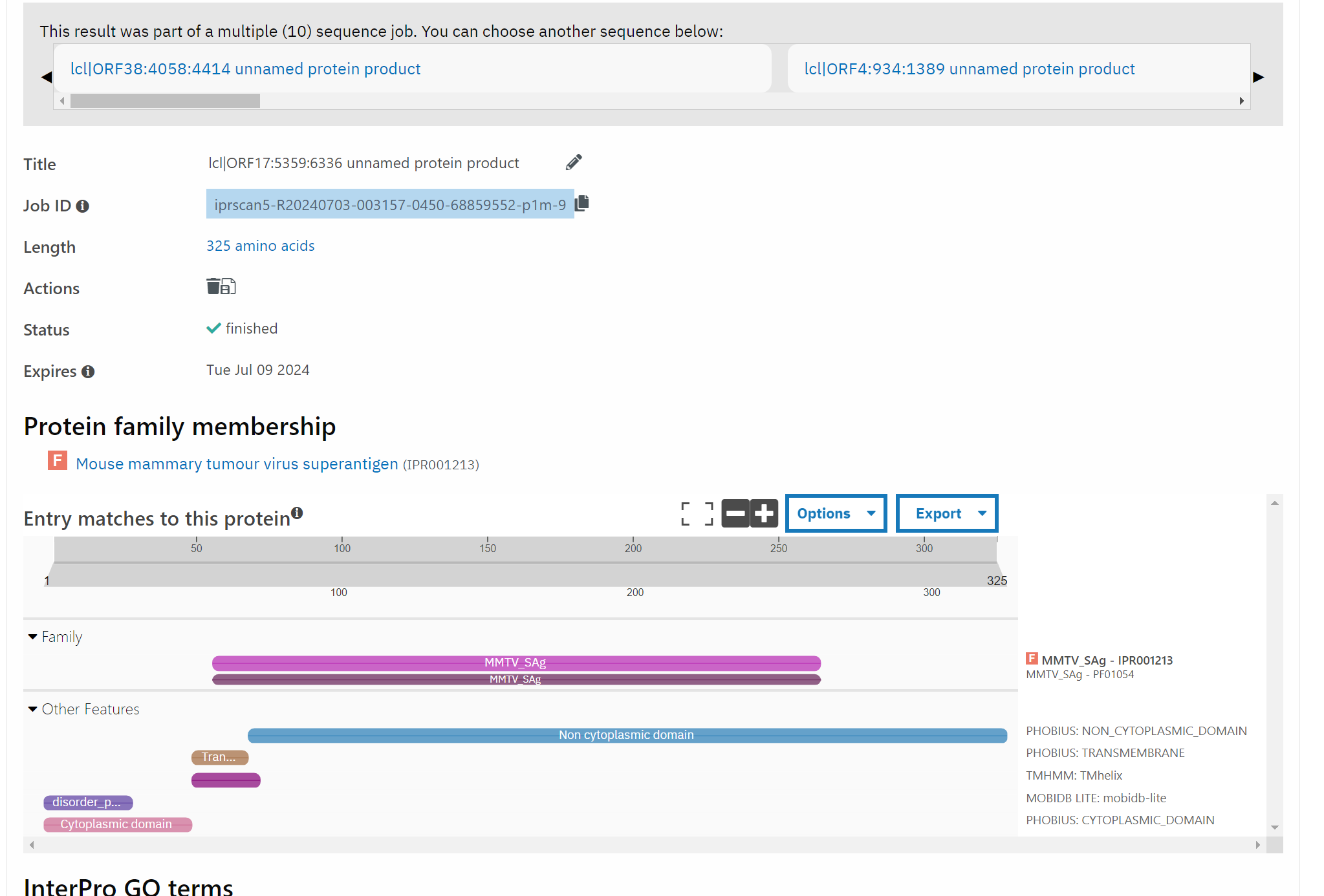
There was no indication that this was a purposeful product of the experiment in the original publishing paper (see BioProject page here). Again, we need n > 1!
So, I then grabbed all the runs from this BioProject from logan, and did a DIAMOND alignment to see if there are any other contigs similar to this one where there would be a mix of MMTV and PV sequences. Started with spot checking - there was a 9kb contig SRR830321_224 (name has changed now due to update to Logan v1.1, this investigation was performed on v1.0) that contained PV E1, E7, E6 and E2 PV domains, a bacterial MFS transporter (might be part of the backbone), and also the MMTV SAg, and RAS GTPase regions.
There was also a contig SRR830327_9, that contained almost a complete PV genome with addition of RAS and MFS, but no MMTV.
To try to get some more context, I reassembled some of these libraries with SPAdes, and got a circular contig that has all the PV genes on the same contig as expected plasmid vector elements - some other summaries below:
| CONTIG NAME_LIBRARY | CONTAINS (BLASTp, not comprehensive) |
|---|---|
| NODE_1_length_8802_cov_9.743642_SRR83010 | BPV1 L1, L2, E1, E2, E4, E5, E6, E7, E8, MMTV Sag, Tet resistance MFS, murine transforming protein p21 |
| NODE_2_length_7118_cov_6.442061_SRR830295 | BPV1 L1, L2, E1, E2, E4, E5, E6, E7, E8, MMTV Sag |
| NODE_2_length_7214_cov_7.198858_SRR830296 | BPV1 L1, L2, E1, E2, E4, E5, E6, E7, E8, MMTV Sag |
| NODE_2_length_7132_cov_13.870686_SRR830297 | BPV1 L1, L2, E1, E2, E4, E5, E6, E7, E8, MMTV Sag |
| NODE_1_length_8730_cov_9.754858_SRR830298 | BPV1 L1, L2, E1, E2, E4, E5, E6, E7, E8, MMTV Sag, Tet resistance MFS murine transforming protein p21 |
n > 1!
I did a quick visualization for NODE_1_length_8802_cov_9.743642_SRR830310 based on InterProScan hidden Markov models, compared against the HPV16 genome organization:
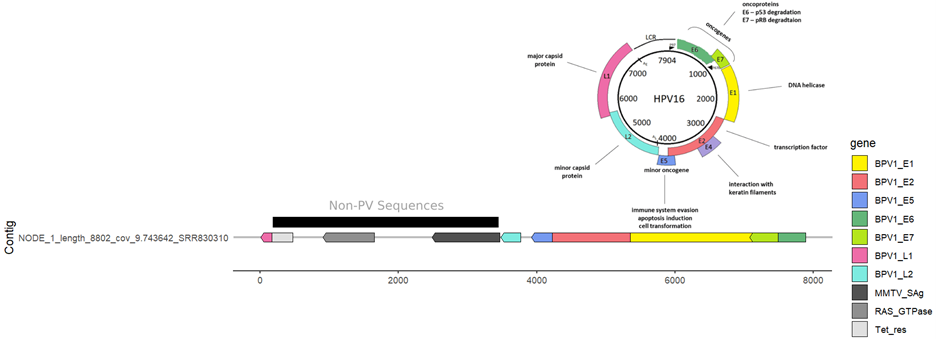
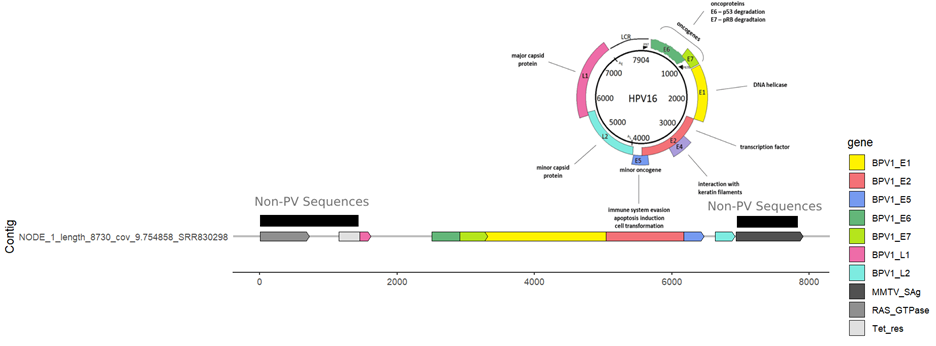
Seems pretty solid to me! Something is weird about these vectors - some of the PV sequences match to BPV and HPV, which makes me think perhaps it has to do with the fetal bovine serum, or human contamination from the experiment. Pretty cool to be able to pick this up from a Logan run (I emailed the submitters, but did not receive a response).
Whew! All in a days work (more like 2 days, but…).
July 25, 2025 JS