So you think you can be a dry lab biologist
(spoilers) You can!
This is a post that will be adapted for the Wiki of my lab sometime in the near future.
The most common question I get during the start of the new year from bright-eyed soon-to-be graduate students is: “I only have wet lab experience, is it possible for me to do good work in a dry lab?”. My short answer is always “Yes!”, and this is applicable to any squishy wet lab biologists reading this right now as well.
I feel like I have a certain level of qualification to talk about this subject, as I am pretty much as described above. My bachelor’s degree was almost entirely wet lab based, learning for example, the both the theory behind and applying hands-on PCR and cloning. The only true formal experience I had learning coding was about 8 different “here’s a barebones introduction to R/Unix” at the beginning of various classes. I think I checked out around the 3rd time I had the same tutorial (load packages, assign variables yada yada). That’s not to say that these classes were not useful - they are useful if you have very minimal coding experience and are just starting out. But, to actually really learn to do bioinformatics well is a whole ‘nother beast.
So, how to get a few steps further than basic tutorials?
The main piece of advice I give here is do a project. Find a question you are interested in, no matter how trivial and just go and try to figure it out. This is sometimes hard if you’re not used to the loop of scientific thinking, but a few things off the top of my head that I would investigate if I had the capacity to: 1) Using a tool like logan-search, has the genetic sequence of salmon viruses (or insert your system of interest) changed over the last 2 decades, or where they appear geographically? Can you track the introduction of viruses in and out of certain oceans? 2) This paper provides a solid starting hypothesis, of given a certain geographical area, can you track changes in beneficial bacteria or pathogenic ones? 3) Using Serratus, characterize a novel virus using all available information. These are just some simple examples, but you should really pursue something you are interested in (and to be honest, the above are probably a bit complicated to start with but I digress).
So, you have a question. What now?
In my opinion, the most important thing now is to really get to know what you are working with, and what tools you have to manipulate them. Below is a collection of resources and tools that I have found useful, your mileage may vary. If there is a term that you’re unfamiliar with, I encourage you to search it up and try to gain a slight-more-than surface-level understanding of the concept, data format, or process, which will pay dividends further down the road.
What am I working with?
There are a lot of file formats and terms in bioinformatics that documentation will just expect you to know, so hopefully my candid descriptions here help in that aspect.
File formats
FASTA
In bioinformatics, you’re most often working with, at a fundamental level, sequencing data in some kind of FASTA format. It is imperative that whenever you work with sequencing data you understand where it is coming from. A metagenomic versus genomic versus transcriptomic sequencing library will tell you very different things about what you are looking at and if it is important or not. You should also know how the data is generated, roughly anyways. PolyA enrichment? PCR amplification beforehand? etc.
In terms of the file itself, NCBI has a really nice write-up here. TLDR: information about the following sequence goes in a line that starts with “>”, with the sequence on the line directly after. It can be in one line format (line with “>” immediately followed by a single long line of sequences) or multi-line format (line with “>”, and n lines of sequences afterwards). While the NCBI article does not explicitly mention it, FASTA files can also be translated to amino acids. It is generally a good idea to shove as much relevant information into the header as possible, as it’ll help you keep track of which is which, and is it relatively simple to extract that metadata for future use - this is where some foresight is useful based on your specific question.
An extension of the FASTA is an multiple sequence alignment (MSA). Wikipedia has a surprisingly nice article on this here, and a more tutorial-like one here.
A bit less common in my work are SAM/BAM files - but good to know about the existence of in general.
Tables, unlimited tables, but no tables (blastfmt6s, domtbls)
There are two major table formats that I work with almost daily - the BLAST 6 format (sometimes blast6fmt, blast6, blast6out….), and the HMMER domtbl (domain table). Sometimes headers won’t be included in the outputs, so it is a good idea to roughly know what you are looking at - this is a nice resource for understanding blastfmt6 from the metagenomics wiki, and you can often set what parameters you want output on your own, so remember to keep track of those. The HMMER User’s Guide is pretty much the go-to for anything HMMER related, so refer to that for formats.
Other tables will also come up, such as BED files, which can be used to annotate regions of FASTA sequences, to either extract/remove/trim them.
“Manipulation” programs
This is a category of programs that can be used to massage the above file formats into the form that you want. In Unix, I looooove seqkit. The FASTA to tab and vice versa is really nice for subsetting or filtering data, or even switching from multi- to one-line format. Splitting can be useful for sampling, renaming if there are dupe names, looking for duplicate sequences etc… In general, if it has to do with changing the format of a FASTA file, seqkit’s got you covered. Not specific to bioinformatics, but learning to use the suite of sed/awk/grep are also pretty invaluable, but is a huge can of worms best not explained by me but by Matt Probert here. EMBOSS is another such Swiss army knife-type program - I usually use it for translation nucleotides into amino acids, and it contains a suite of different assembly methods, trimming, filtering, etc. This is set up on the Donnelly Cluster already, and should be accessible with the right bashrc settings.
If you are working with short reads, or need to re-assemble a library, my go to is SPAdes, which comes with a few different versions for different assembly uses. It’s good background to generally know how assemblers work in principal, as well.
Often, you will need to cluster sequences based on nucleotide or amino acid identity to reduce redundancy - e.g., you don’t want a FASTA file where 95% of the sequences are 1 base off from each other. It won’t be very helpful in making trees, or accurately representing the diversity contains within a set. Obviously this is not always true, as sometime’s it’s useful to have those raw numbers, but a lot of the time, redundancy should be removed. Use your best judgement; what is the question you are trying to answer? Sometimes, clustering is also useful in seeing the level of overall diversity - how many clusters at 90, 80, 70% identity? Typically, I use usearch to do this clustering.
To align sequences to generate a MSA, I use MUSCLE, which is quite simple to use, with super5.
I do the majority of my analyses in R, especially on tables. I never load R without tidyverse. This is a large package with a lot of functionality, but at the most basic level you will need to know how to use read.table, probably filter and basic R commands like adding/manipulating/moving columns (which is best done with lots of Google-fu and trial and error).
“Experiment” programs
A lot of bioinformatics meat lives here. These are the programs that you use to try to answer biological questions. What sequences are similar to other sequences? By how much? Which are new, which are known?
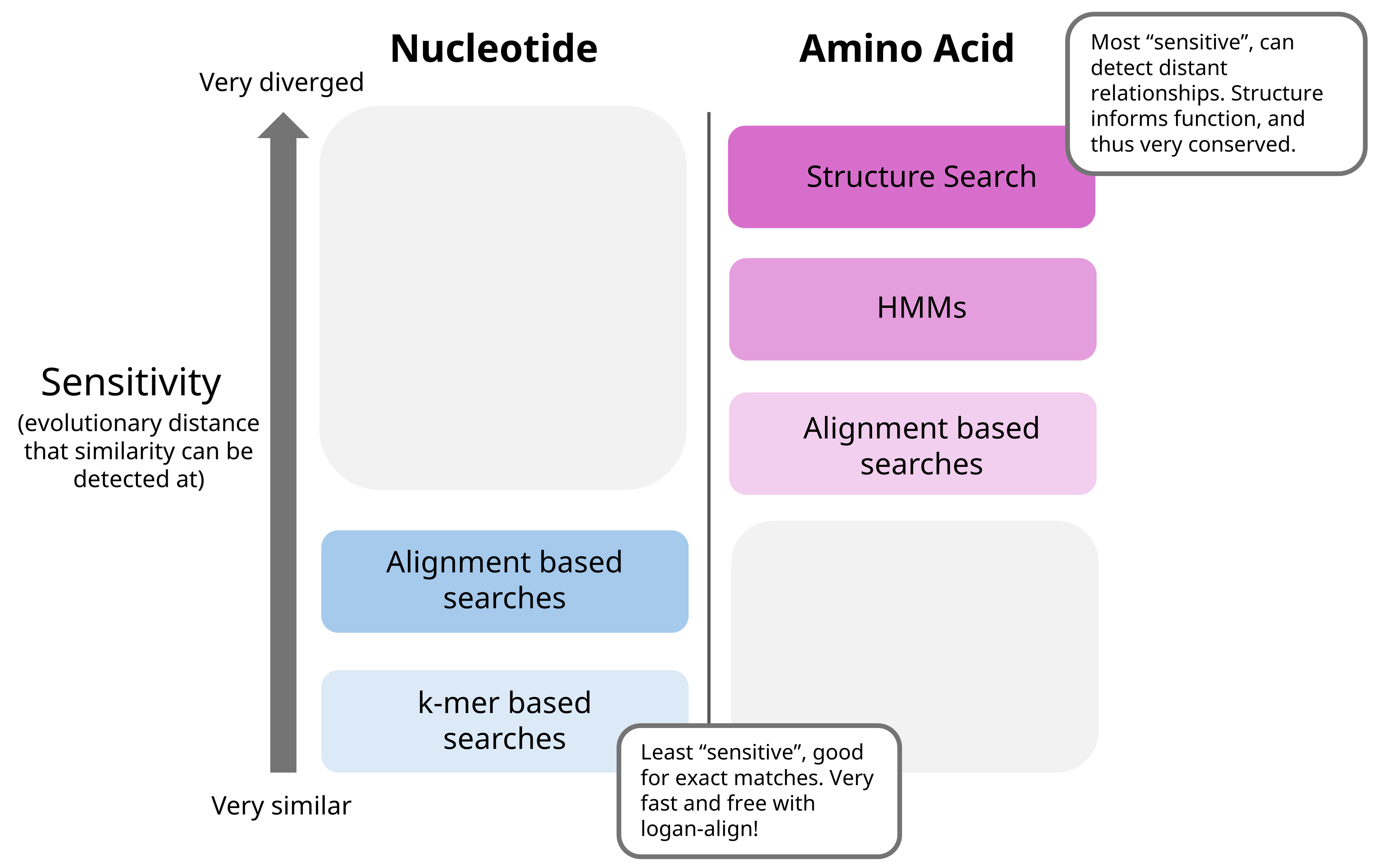
At the most fundamental level, there are the BLAST-like programs - know how these work, generally (BLOSUMs, alignments). The one most commonly used in my lab is DIAMOND, for amino acid searches or translated searches against a provided database (either your own, or blastnr). The documentation is pretty solid, so take a read through it at some point before using. DIAMOND is quite a bit faster than the usual blastp search from NCBI toolkits, so I suggest using this when possible. For nucleotide searches, the BLAST command line apps work well, see here for documentation. If you are working on the Donnelly Centre cluster, this has already been set up in a shared folder. It will take roughly forever to download and build the database from scratch, so this is not recommended (unless it is super out of date).
Another iteration of such searches, based on another concept, hidden Markov models, is a program mentioned above called HMMER. The general concept is different from BLAST searches, and they can usually detect things that are a bit more evolutionarily distant. HMMER requires your query sequences, as well as a HMM to search against (e.g., the pattern that you want to find in your queries). The User’s Guide has a good tutorial on how it works, but you start with a MSA, and convert to the appropriate format to work with HMMER. Using the same concept of HMMs, the Pfam online tool is quite good for searching through all recorded HMMs of various protein domains, if you want to see what is on an amino acid sequence of interest or identify what it might be.
I generate phylogenetic trees, which are paramount in understanding evolutionary relationships, in two main ways 1) I really really need to see this tree right now or I’ll explode (the faster and dirtier way), using this online tool, and the FastTree option. The other way is 2) This tree will take a while, but have nice bootstrap values and inspire confidence, through IQ-TREE 2, and 1k bootstrap iterations (read more on what bootstraps are here).
Alphafold is another classic, which can be used to speedily and accurately predict protein structures from amino acid sequences. This can be extended to structure search programs like Foldseek, which is another step above HMMs in looking for distantly related proteins (and use PyMol to visualize these things!).
In general, I think of these programs as such in terms of sensitivity:
“Visualization” programs
ggplot2 is my best friend. I would say it is highly worth it to get a basic understanding of it’s syntax - it’s been invaluable in whipping up a quick graph to visualize interim results or investigate discrepancies in data. I like to use R Graph Gallery as a starting point for my graphs, then adjust for aesthetics and formats from there. In a similar vein I use ggtree for visualizing phylogenetic trees. There’s a bit of a learning curve - a faster, more interactive option is iTOL, but it isn’t quite as powerful and a bit more of a hassle. Photopea and …Powerpoint… are good for working with .svgs and making figures.
Jalview is my go-to for visualizing MSAs (look at the overview window in the View tab, change the colors to Taylor, zoom in and out…). There are a few good reasons as to why we visualize MSAs - it gives information on where in the sequences things are conserved (or not conserved), if there are any gaps (e.g., do some sequences have and insertion or deletion compared to the rest), do ends need to be trimmed before generating an HMM? And in general, it is good to look at your data manually to make sure everything looks good!
You can use the macro below in Powerpoint to export some real high quality figures:
Option Explicit
' These items govern the export format and resolution
' Edit them as needed
' If you use a Mac, substitute colons for backslashes as path separators
' What image format is desired?
Const ExportFormat As String = "PNG" ' change to "PNG" or whatever's needed
' Image width and height
' Make sure they're proportional to the slide's dimensions
Const ExportWidth As Long = 8192
Const ExportHeight As Long = 4607.954
' In what folder should we put the images?
' MUST end with a \ character
Const ExportFolder As String = "C:\Users\path\to\destination\"
' What should we name the images?
' Slide number will be appended to this base name
Const BaseName As String = "slide_image_prefix"
Sub ExportSlides()
Dim oSl As Slide
For Each oSl In ActivePresentation.Slides
oSl.Export ExportFolder & BaseName & Format(oSl.SlideIndex, "0000") _
& "." & ExportFormat, _
ExportFormat, ExportWidth, ExportHeight
Next ' Slide
End Sub
Sub png()
End Sub
Sometimes, the easiest thing to do is just to throw some data in Excel and call it day.
To answer a question
This is the part that tends to be the hardest to convey for me - how do you fit all the pieces of tools, data, and ideas to answer a question? For me, it feels like almost a puzzle: you need to collect all the pieces, and then fit them together in a way that makes sense. First of all, the list above is far far far from comprehensive. For instance, if I wanted to take a crack at seeing if a specific Papillomavirus species was around in 2015 but not 2025, I’d do a search with logan-search. Export the table, look at it in R. Hey, there might be some weird pattern happening, for example, it might be present only in certain years but not others, which I might want to investigate. Maybe the Logan contigs aren’t long enough to give me good information on the genome. I’d then have to download the raw reads with SRA Toolkit, then reassemble it, maybe against a reference genome using Bowtie. Maybe I want the general metadata for all SRA entries to compare, so I’ll need to query an SQL table for a certain range of years or library types. Maybe I’m now only interested submissions from a certain institution, so I would grep for those fields, get the accessions, and filter for those contigs?
Overall, my advice here would be to get as far as you can, use alot of Google, and once you start getting to the point where it’s become not very productive to sit and stare and think, ask for help. It’s a bit of a muscle, and I promise it gets easier with time.
More formal classes and learning
Sometimes, a more structured approach works better for learning the very basics. Here are some great, free resources to get started:
- Data Science: R Basics
- CS50’s Introduction to Programming with Python
- BIOL525D: Bioinformatics for Evolutionary Biology and Conservation
- Tidvyerse Basics
- BIO300: The Analysis of Biological Data - this one is more biostats focused, but there are some good tidbits on using R for this purpose.
- Serratus Tutorials - a really good place to start, step by step.
I also have some other short write ups, that use these programs in a few ways - might be helpful to check out.
August 20, 2025 JS